医療機器を市場に流通させるために必要なこと:医療機器開発のための薬機法解説(2)(1/3 ページ)
2014年11月に施行された医薬品医療機器等法、略して「薬機法」について、医療機器開発の観点から解説していく本連載。第2回は、医療機器を市場に流通させる際の規制などについ取り上げる。
1)前回まで
連載第1回では、医薬品医療機器等法の全体像についてのお話と、医療機器とは何か? というお話をしました。
医薬品医療機器等法は正式な名前を「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」といい、医療機器の他、医薬品や化粧品などの安全性確保などのために必要な規制を行う法律です。
長い名前なので、短くすると「医薬品医療機器等法」、さらに略して「薬機法」とも呼ばれています。
医療機器に関する法律は、以前は「薬事法」という名前でしたが、平成26(2014)年の大きな改正の際に名前が変更されました。
この医薬品医療機器等法の第二条第四項では、医療機器の定義として「人若しくは動物の疾病の診断、治療若しくは予防に使用されること、又は人若しくは動物の身体の構造若しくは機能に影響を及ぼすことが目的とされている機械器具等(再生医療等製品を除く。)であつて、政令で定めるものをいう。」と規定しています。
第2回はもう少し具体的な内容に踏み込んで、医療機器等を市場に流通させるために必要なことについて、触れていきたいと思います。
2)市場流通させるために必要な規制
医薬品医療機器等法では、製品を市場に流通させるに当たって、品質、有効性、安全性の確保のために、2つの側面から規制を行っています。
1つ目は、業者側の規制です。製造や製造販売などの行為をなりわい、すなわち「業」として行うことができるということで、業者は、取扱品目や業態にあわせて許可や登録、届け出を行う必要があります。
これをしないと、製品に対する承認などが取得できたとしても、市場流通はできません。どのような業態に対し許可、届け出などが必要であるのかは品目ごとに異なりますが、以下のような種類があります。
2つ目は、「製品」に対する規制です。製品の品目や種別(医療機器の場合は、そのリスクによって数種に分類されています)によって、それぞれ承認、許可、届け出などの手続きが必要になります。
これにより、「その製品が市場で流通可能であると厚生労働省が認めた」ということになります。
このように「業態」と「製品」の両方で、必要な手続きを経てはじめて、製品を市場に流通させることができます。
3)医療機器における業態と許認可について
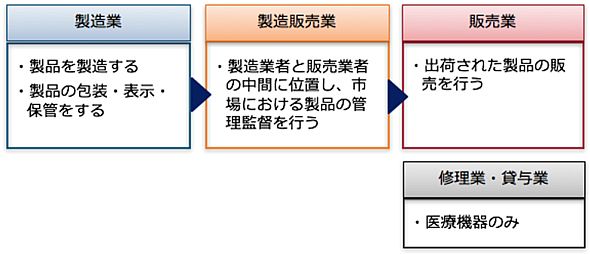
前項で示した「製造業」「製造販売業」「販売業」、そして医療機器独自の業態である「修理業」「貸与業」について、もう少し細かく見てみましょう。
「修理業」は文字通り、医療機器の修理を行う業態のことで、医薬品医療機器等法第四十条の二(医療機器の修理業の許可)において定められた通り、許可制がとられています。
「販売業」と「貸与業」は、出荷された製品の販売や貸与を行う業態のことで、扱う製品の分類によって許可制のものと届け出制のものがあります。
使用リスクに応じた医療機器分類の中で、リスクの高い「高度管理医療機器」などは許可制となっており、リスクの比較的低い「管理医療機器」などは届け出制となっています。
なお、「貸与業」については、薬事法時代には「賃貸業」と呼ばれていましたが、医薬品医療機器等法では名称が「貸与業」に変更され、業として無償で貸与を行う場合も許可、届け出の対象となりました。
「製造販売業」は、薬事法時代の平成17年改正(2005年4月施行)の際に誕生しました。
名前のイメージからすると、製造業、販売業の両方の役割ができるように感じられますが、この製造販売業は、製造業者と販売業者の中間に位置し、安全対策、表示なども含めた製品の最終責任を負う立場のことです。
医療機器製造販売業は医薬品医療機器等法第二十三条の二(製造販売業の許可)において第一種から第三種の3つに区分されており、それぞれ許可制となっています。
最後に「製造業」ですが、設計、組み立て、滅菌、保管など、製品の製造工程を行う業態です。
薬事法時代に許可制がとられていた製造業は、医薬品医療機器等法になってから「登録制」へ移行し、手続きが簡素化されました。
医薬品医療機器等法施行規則第百十四条の八(製造業の登録を受ける製造所の製造工程)において、登録の対象となる工程が定められています。
それぞれの業態についての詳しい要件など、次回以降もう少し掘り下げて行きたいと思いますが、次項にあげる品質基準や安全基準にもそれぞれ適合していく必要があります。
Copyright © ITmedia, Inc. All Rights Reserved.